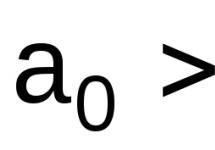Термодинамическая система – тело или группа тел, находящихся во взаимодействии, мысленно или реально обособленные от окружающей среды.
Гомогенная система – система, внутри которой нет поверхностей, разделяющих отличающиеся по свойствам части системы (фазы).
Гетерогенная система – система, внутри которой присутствуют поверхности, разделяющие отличающиеся по свойствам части системы.
Фаза – совокупность гомогенных частей гетерогенной системы, одинаковых по физическим и химическим свойствам, отделенная от других частей системы видимыми поверхностями раздела.
Изолированная система – система, которая не обменивается с окружающей средой ни веществом, ни энергией.
Закрытая система – система, которая обменивается с окружающей средой энергией, но не обменивается веществом.
Открытая система – система, которая обменивается с окружающей средой и веществом, и энергией.
Параметры состояния – величины, характеризующие какое-либо макроскопическое свойство рассматриваемой системы.
Термодинамический процесс – всякое изменение термодинамического состояния системы (изменения хотя бы одного параметра состояния).
Обратимый процесс – процесс, допускающий возможность возвращения системы в исходное состояние без того, чтобы в окружающей среде остались какие-либо изменения.
Равновесный процесс – процесс, при котором система проходит через непрерывный ряд состояний, бесконечно близких к состоянию равновесия. Характерные особенности равновесного процесса:
1) бесконечно малая разность действующих и противодействующих сил: F ex – F in > 0;
2) совершение системой в прямом процессе максимальной работы |W | = max;
3) бесконечно медленное течение процесса, связанное с бесконечно малой разностью действующих сил и бесконечно большим числом промежуточных состояний t > ?.
Самопроизвольный процесс – процесс, который может протекать без затраты работы извне, причем в результате может быть получена работа в количестве, пропорциональном произошедшему изменению состояния системы. Самопроизвольный процесс может протекать обратимо или необратимо.
Несамопроизвольный процесс – процесс, для протекания которого требуется затрата работы извне в количестве, пропорциональном производимому изменению состояния системы.
Энергия – мера способности системы совершать работу; общая качественная мера движения и взаимодействия материи. Энергия является неотъемлемым свойством материи. Различают потенциальную энергию, обусловленную положением тела в поле некоторых сил, и кинетическую энергию, обусловленную изменением положения тела в пространстве.
Внутренняя энергия системы U – сумма кинетической и потенциальной энергии всех частиц, составляющих систему. Можно также определить внутреннюю энергию системы как ее полную энергию за вычетом кинетической и потенциальной энергии системы как целого. [U ] = Дж.
Теплота Q – форма передачи энергии путем неупорядоченного движения молекул, путем хаотических столкновений молекул двух соприкасающихся тел, т. е. путем теплопроводности (и одновременно путем излучения). Q > 0, если система получает теплоту из окружающей среды. [Q ] = Дж.
Работа W – форма передачи энергии путем упорядоченного движения частиц (макроскопических масс) под действием каких-либо сил. W > 0, если окружающая среда совершает работу над системой. [W] = Дж.
Вся работа делится на механическую работу расширения (или сжатия) и прочие виды работы (полезная работа): ?W = -pdV + ?W?.
Стандартное состояние твердых и жидких веществ – устойчивое состояние чистого вещества при данной температуре под давлением р = 1атм.
Стандартное состоянии чистого газа – состояние газа, подчиняющееся уравнению состояния идеального газа при давлении 1 атм.
Стандартные величины – величины, определенные для веществ, находящихся в стандартном состоянии (обозначаются надстрочным индексом 0).
1.1. Первое начало термодинамики
Энергия неуничтожаема и несотворяема; она может только переходить из одной формы в другую в эквивалентных соотношениях.
Первое начало термодинамики представляет собой постулат – оно не может быть доказано логическим путем или выведено из каких-либо более общих положений.
Первое начало термодинамики устанавливает соотношение между теплотой Q, работой W и изменением внутренней энергии системы?U .
Изолированная система
Внутренняя энергия изолированной системы остается постоянной.
U = const или dU = 0Закрытая система
Изменение внутренней энергии закрытой системы совершается за счет теплоты, сообщенной системе, и/или работы, совершенной над системой.
?U =Q +W или dU = ? Q + ?WОткрытая система
Изменение внутренней энергии открытой системы совершается за счет теплоты, сообщенной системе, и/или работы, совершенной над системой, а также за счет изменения массы системы.
?U =Q +W + ?U m или dU = ? Q + ?W + i ?U i dn iВнутренняя энергия является функцией состояния; это означает, что изменение внутренней энергии?U не зависит от пути перехода системы из состояния 1 в состояние 2 и равно разности величин внутренней энергии U 2 и U 1 в этих состояниях:
?U =U 2 – U 1Для некоторого процесса:
?U = ?(v i U i) npoд – ?(v i U i) исх1.2. Применение первого начала термодинамики к гомогенным однокомпонентным закрытым системам
Изохорный процесс (V = const; ?V = 0)В простейшем случае – полезная работа не совершается.
dU = ? Q + ?W = ? Q – pdV dU = ?Q v = C V dT = nC V dTВсе количество теплоты, полученное системой, идет на изменение внутренней энергии.
– теплоемкость при постоянном объеме, т. е. количество теплоты, необходимое для повышения температуры системы на один градус при постоянном объеме. [С V ] = Дж/град.
C V – мольная теплоемкость при постоянном объеме, Дж/(моль? град). Для идеальных газов:
C V = 2 / 3 R – одноатомный газ;
C V = 5 / 2 R – двухатомный газ.
Изобарный процесс (Р = const) dU = ? Q + ?W = ?Q – pdV ?Q p = dU + pdV = d(U + pV) = dHH = U + pV – энтальпия – функция состояния системы.
?Н = ?(? i U i) прод – ?(? i U i) исх?Q p = dU + pdV =dH = C p dT – тепловой эффект изобарного процесса равен изменению энтальпии системы.

– теплоемкость при постоянном давлении. [С ] = Дж/град.
C р – мольная теплоемкость при постоянном давлении, Дж/(моль? град).
Для идеальных газов: C р = C V + R; C р, C V = [Дж/(моль К)].
Тепловой эффект (теплота) химической реакции – количество теплоты, выделившейся либо поглотившейся в ходе реакции при постоянной температуре.
Q v = ?U V Q p = ?U p Зависимость теплового эффекта реакции от температуры. Закон КирхгоффаТемпературный коэффициент теплового эффекта химической реакции равен изменению теплоемкости системы в ходе реакции.
Закон Кирхгоффа:
Для химического процесса изменение теплоемкости задается изменением состава системы:
?С р = ?(? i C p,i) прод – ?(? i C p,i) исх или?C V = ?(? i C V,i) прод – ?(? i C V,i) исхИнтегральная форма закона Кирхгоффа:
?Н Т2 = ?Н Т1 + ?С р (Т 2 – T 1 ) или?U T2 = ?U Ti + ?С V (Т 2 – T 1 )1.3. Второе начало термодинамики. Энтропия
1) Теплота не может самопроизвольно переходить от менее нагретого тела к более нагретому.
2) Невозможен процесс, единственным результатом которого является превращение теплоты в работу.
3) Существует некоторая функция состояния системы, названная энтропией, изменение которой следующим образом связано с поглощаемой теплотой и температурой системы:
в неравновесном процессе
в равновесном процессе
S – энтропия, Дж/град,
– приведенная теплота.
Статистическая интерпретация энтропииКаждому состоянию системы приписывается термодинамическая вероятность (определяемая как число микросостояний, составляющих данное макросостояние системы), тем большая, чем более неупорядоченным или неопределенным является это состояние. Энтропия – функция состояния, описывающая степень неупорядоченности системы.
S = k lnW – формула Больцмана.
Система стремится самопроизвольно перейти в состояние с максимальной термодинамической вероятностью.
Расчет абсолютной энтропии
Изменение энтропии в ходе химического процесса определяется только видом и состоянием исходных веществ и продуктов реакции и не зависит от пути реакции:
?S = ?(? i S i) прод – ?(? i S i) исхВеличины абсолютной энтропии в стандартных условиях приведены в справочной литературе.
1.4. Термодинамические потенциалы
Потенциал – величина, убыль которой определяет производимую системой работу.
Самопроизвольно могут протекать только те процессы, которые приводят к понижению свободной энергии системы; система приходит в состояние равновесия, когда свободная энергия достигает минимального значения.
F = U – TS – свободная энергия Гельмгольца – изохорно-изотермический потенциал (Дж) – определяет направление и предел самопроизвольного протекания процесса в закрытой системе, находящейся в изохорно-изотермических условиях.
dF = dU – TdS или?F = ?U – T?SG = H – TS = U + pV – TS – свободная энергия Гиббса – изобарно-изотермический потенциал (Дж) – определяет направление и предел самопроизвольного протекания процесса в закрытой системе, находящейся в изобарно-изотермических условиях.
dG = dH – TdS или?G = ?Н – T?S ?G = ?(? i G i) прод – ?(? i G i) исх ?G 0 = ?(? i ?G обр 0) прод – ?(? i ?G обр 0) исх Условия самопроизвольного протекания процессов в закрытых системахИзобарно-изотермические (Р = const, Т = const):
?G < 0, dG < 0Изохорно-изотермические (V = const, Т = const):
?F < 0, dF < 0Термодинамическим равновесием называется такое термодинамическое состояние системы с минимальной свободной энергией, которое при постоянстве внешних условий не изменяется во времени, причем эта неизменяемость не обусловлена каким-либо внешним процессом.
Условия термодинамического равновесия в закрытой системеИзобарно-изотермические (Р = const, Т = const):
?G = 0, dG = 0, d 2 G > 0Изохорно-изотермические (V = const, Т = const):
?F = 0, dF = 0, d 2 F > 0 Уравнения изотермы химической реакции:Для реакции v 1 A 1 + v 2 A 2 + … = v? 1 B 1 + v? 2 B 2 + …

Здесь C i ,p i – концентрации, давления реагирующих веществ в любой момент времени, отличный от состояния равновесия.
Влияние внешних условий на химическое равновесиеПринцип смещения равновесия Ле Шателье-Брауна
Если на систему, находящуюся в состоянии истинного равновесия, оказывается внешнее воздействие, то в системе возникает самопроизвольный процесс, компенсирующий данное воздействие.
Влияние температуры на положение равновесия
Экзотермические реакции: ?Н° < 0 (?U° < 0). Повышение температуры уменьшает величину константы равновесия, т. е. смещает равновесие влево.
Эндотермические реакции: ?Н° > 0 (?U° > 0). Повышение температуры увеличивает величину константы равновесия (смещает равновесие вправо).
2. Фазовые равновесия
Компонент – химически однородная составная часть системы, которая может быть выделена из системы и существовать вне ее. Число независимых компонентов системы равно числу компонентов минус число возможных химических реакций между ними.
Число степеней свободы – число параметров состояния системы, которые могут быть одновременно произвольно изменены в некоторых пределах без изменения числа и природы фаз в системе.
Правило фаз Дж. Гиббса:
Число степеней свободы равновесной термодинамической системы С равно числу независимых компонентов системы К минус число фаз Ф плюс число внешних факторов, влияющих на равновесие: С = К – Ф + n.
Для системы, на которую из внешних факторов влияют только температура и давление, можно записать: С = К – Ф + 2.
Принцип непрерывности – при непрерывном изменении параметров состояния все свойства отдельных фаз изменяются также непрерывно; свойства системы в целом изменяются непрерывно до тех пор, пока не изменится число или природа фаз в системе, что приводит к скачкообразному изменению свойств системы.
Согласно принципу соответствия, на диаграмме состояния системы каждой фазе соответствует часть плоскости – поле фазы. Линии пересечения плоскостей отвечают равновесию между двумя фазами. Всякая точка на диаграмме состояния (т. н. фигуративная точка) отвечает некоторому состоянию системы с определенными значениями параметров состояния.
2.1. Диаграмма состояния воды
К = 1. В системе возможны три фазовых равновесия: между жидкостью и газом (линия ОА), твердым телом и газом (линия ОВ), твердым телом и жидкостью (линия OC). Три кривые имеют точку пересечения О, называемую тройной точкой воды, – отвечают равновесию между тремя фазами и С = 0; три фазы могут находиться в равновесии лишь при строго определенных значениях температуры и давления (для воды тройная точка отвечает состоянию с Р = 6,1 кПа и Т = 273,16 К).

Внутри каждой из областей диаграммы (АОВ, ВOC, АOC) система однофазна; С = 2 (система бивариантна).
На каждой из линий число фаз в системе равно двум, и, согласно правилу фаз, система моновариантна: С = 1 – 2 + 2 = 1, т. е. для каждого значения температуры имеется только одно значение давления.
Влияние давления на температуру фазового перехода описывает уравнение Кла-узиуса – Клапейрона:

V 2 , V 1 – изменение молярного объема вещества при фазовом переходе.
Кривая равновесия «твердое вещество – жидкость» на диаграмме состояния воды наклонена влево, а на диаграммах состояния остальных веществ – вправо, т. к. плотность воды больше, чем плотность льда, т. е. плавление сопровождается уменьшением объема (AV < 0). В этом случае увеличение давления будет понижать температуру фазового перехода «твердое тело – жидкость» (вода – аномальное вещество). Для всех остальных веществ (т. н. нормальные вещества) ?V пл > 0 и, согласно уравнению Клаузиуса-Клапейрона, увеличение давления приводит к повышению температуры плавления.
3. Свойства растворов
3.1. Термодинамика растворов
Раствор – гомогенная система, состоящая из двух или более компонентов, состав которой может непрерывно изменяться в некоторых пределах без скачкообразного изменения ее свойств.
Диффузия в растворах
Диффузия – самопроизвольный процесс выравнивания концентрации вещества в растворе за счет теплового движения его молекул или атомов.
Закон Фика: количество вещества, диффундирующее за единицу времени через единицу площади поверхности пропорционально градиенту его концентрации:
где j – диффузионный поток; D – коэффициент диффузии.
Уравнение Эйнштейна-Смолуховского:

где? – вязкость среды; R – радиус диффундирующих частиц.
Растворимость газов в газахЗакон Дальтона: общее давление газовой смеси равно сумме парциальных давлений всех входящих в нее газов:
Р общ = ?p i и pi = xi Р общЗакон Генри-Дальтона: растворимость газа в жидкости прямо пропорциональна его давлению над жидкостью: C i = kp i , где C i – концентрация раствора газа в жидкости; k – коэффициент пропорциональности, зависящий от природы газа.
Как правило, при растворении газа в жидкости выделяется теплота (к < 0), поэтому с повышением температуры растворимость уменьшается.
Формула Сеченова:
X =Х 0 е -kС элгде X и Х 0 – растворимость газа в чистом растворителе и растворе электролита с концентрацией С.
3.2. Коллигативные свойства растворов неэлектролитов
Коллигативными (коллективными) называются свойства растворов относительно свойств растворителя, зависящие главным образом от числа растворенных частиц.
Давление насыщенного пара разбавленных растворовПар, находящийся в равновесии с жидкостью, называется насыщенным. Давление такого пара р 0 называется давлением или упругостью насыщенного пара чистого растворителя.
Первый закон Рауля. Парциальное давление насыщенного пара компонента раствора прямо пропорционально его мольной доле в растворе, причем коэффициент пропорциональности равен давлению насыщенного пара над чистым компонентом:
p i = p i 0 x iДля бинарного раствора, состоящего из компонентов А и В: относительное понижение давления пара растворителя над раствором равно мольной доле растворенного вещества и не зависит от природы растворенного вещества:

Растворы, для которых выполняется закон Рауля, называют идеальными растворами.
Давление пара идеальных и реальных растворов
Если компоненты бинарного (состоящего из двух компонентов) раствора летучи, то пар над раствором будет содержать оба компонента. Общее Состав, мол. доли в (х в) давление пара:
p = p A 0 x A + p B 0 x B = p A 0 (1 – x B) + p B 0 x B = p A 0 – x B (p A 0 – p B 0)
Если молекулы данного компонента взаимодействуют друг с другом сильнее, чем с молекулами другого компонента, то истинные парциальные давления паров над смесью будут больше, чем вычисленные по первому закону Рауля (положительные отклонения, ?Н тв > 0). Если же однородные частицы взаимодействуют друг с другом слабее, чем разнородные, парциальные давления паров компонентов будут меньше вычисленных (отрицательные отклонения, ?H раств < 0).
Температура кристаллизации разбавленных растворовВторой закон Рауля. Понижение температуры замерзания раствора?Т зам прямо пропорционально моляльной концентрации раствора: ?T зам = Т 0 – Т = КС m , где Т 0 – температура замерзания чистого растворителя; Т – температура замерзания раствора; К – криоскопическая постоянная растворителя, град/кг моль,

Т 0 2 – температура замерзания растворителя; М – молекулярная масса растворителя, ?Н пл – мольная теплота плавления растворителя.
Температура кипения разбавленных растворовТемпература кипения – температура, при которой давление насыщенного пара становится равным внешнему давлению.
Повышение температуры кипения растворов нелетучих веществ?Т К = Т к – Т к 0 пропорционально понижению давления насыщенного пара и прямо пропорционально моляльной концентрации раствора: ?Т кип = ЕС m , где Е – эбулиоскопическая постоянная растворителя, град/кг моль,
 Осмотическое давление разбавленных растворов
Осмотическое давление разбавленных растворов
Осмос – преимущественно одностороннее прохождение молекул растворителя через полупроницаемую мембрану в раствор или молекул растворителя из раствора с меньшей концентрацией в раствор с большей концентрацией.
Давление, которое необходимо приложить к раствору, чтобы предотвратить перемещение растворителя в раствор через мембрану, разделяющую раствор и чистый растворитель, численно равно осмотическому давлению? (Па).
Принцип Вант-Гоффа: осмотическое давление идеального раствора равно тому давлению, которое оказывало бы растворенное вещество, если бы оно, находясь в газообразном состоянии при той же температуре, занимало бы тот же объем, который занимает раствор: ? = CRT.
Изотонические растворы – два раствора с одинаковым осмотическим давлением (? 1 = ? 2).
Гипертонический раствор – раствор, осмотическое давление которого больше, чем у другого (? 1 > ? 2).
Гипотонический раствор – раствор, осмотическое давление которого меньше, чем у другого (? 1 < ? 2).
3.3. Растворы электролитов
Степень диссоциации? – отношение числа молекул n, распавшихся на ионы, к общему числу молекул N:
Изотонический коэффициент i Ван-Гоффа – отношение фактического числа частиц в растворе электролита к числу частиц этого раствора без учета диссоциации.
Если из N молекул продиссоциировало n, причем каждая молекула распалась на? ионов, то

Для неэлектролитов i = 1.
Для электролитов 1 < i ? ?.
3.4. Коллигативные свойства растворов электролитов:
 Теория электролитической диссоциации Аррениуса
Теория электролитической диссоциации Аррениуса
1. Электролиты в растворах распадаются на ионы – диссоциируют.
2. Диссоциация является обратимым равновесным процессом.
3. Силы взаимодействия ионов с молекулами растворителя и друг с другом малы (т. е. растворы являются идеальными).
Диссоциация электролитов в растворе происходит под действием полярных молекул растворителя; наличие ионов в растворе предопределяет его электропроводность.
По величине степени диссоциации электролиты подразделяются на три группы: сильные (? ? 0,7), средней силы (0,3 < ? < 0,7) и слабые (? ? 0,3).
Слабые электролиты. Константа диссоциацииДля некоторого электролита, распадающегося в растворе на ионы в соответствии с уравнением:
А а В b - аА x- + bВ y+
Для бинарного электролита:

– закон разбавления Оствальда: степень диссоциации слабого электролита возрастает с разбавлением раствора.
Активность растворенного вещества – эмпирическая величина, заменяющая концентрацию, – активность (эффективная концентрация) а, связанная с концентрацией через коэффициент активности f , который является мерой отклонения свойств реального раствора от идеального:
а = fC; а + = f + С + ; а_ = f_C_.Для бинарного электролита:
– средняя активность электролита;
– средний коэффициент активности.
Предельный закон Дебая-Хюккеля для бинарного электролита: lg f = -0,51z 2 I ?, где z – заряд иона, для которого рассчитывается коэффициент активности;
I – ионная сила раствора I = 0,5?(С i r i 2).
4. Электропроводность растворов электролитов
Проводники I рода – металлы и их расплавы, в которых электричество переносится электронами.
Проводники II рода – растворы и расплавы электролитов с ионным типом проводимости.
Электрический ток есть упорядоченное перемещение заряженных частиц.
Всякий проводник, по которому течет ток, представляет для него определенное сопротивление R, которое, согласно закону Ома, прямо пропорционально длине проводника l и обратно пропорционально площади сечения S; коэффициентом пропорциональности является удельное сопротивление материала? – сопротивление проводника, имеющего длину 1 см и сечение 1 см 2:

Величина W, обратная сопротивлению, называется электропроводностью – количественной меры способности раствора электролита проводить электрический ток.

Удельная электропроводность ?(к) – электропроводность проводника I рода длиной 1 м с площадью поперечного сечения 1 м 2 или электропроводность 1 м 3 (1 см 3) раствора электролита (проводника II рода) при расстоянии между электродами 1 м (1 см) и площади электродов 1 м 2 (1 см 2).
Молярная электропроводность раствора) ? – электропроводность раствора, содержащего 1 моль растворенного вещества и помещенного между электродами, расположенными на расстоянии 1 см друг от друга.

Молярная электропроводность как сильных, так и слабых электролитов увеличивается с уменьшением концентрации (т. е. с увеличением разведения раствора V = 1 / C), достигая некоторого предельного значения? 0 (? ?), называемого молярной электропроводностью при бесконечном разведении.
Для бинарного электролита с однозарядными ионами при постоянной температуре и напряженности поля 1 В м -1:
? = ?F(u + + и?),где F – число Фарадея; и + , и? – абсолютные подвижности (м 2 В -1 с -1) катиона и аниона – скорости движения данных ионов в стандартных условиях, при разности потенциалов в 1В на 1 м длины раствора.
? + = Fu + ; ?? = Fu?,где? + , ?? – подвижности катиона и аниона, Ом м 2 моль -1 (Ом см 2 моль -1).
? = ?(? + + ??)Для сильных электролитов? ?1 и ? = ? + + ??
При бесконечном разбавлении раствора (V > ?, ? + > ? ? + , ?? > ? ? ?, ? > 1) как для сильного, так и для слабого электролитов? ? = ? ? + – ? ? ? – закон Кольрауша: молярная электропроводность при бесконечном разведении равна сумме электролитических подвижностей? ? + , ? ? ? катиона и аниона данного электролита.
Ионы Н + и OH? обладают аномально высокой подвижностью, что связано с особым механизмом переноса заряда этими ионами – эстафетным механизмом. Между ионами гидроксония Н 3 O + и молекулами воды, а также между молекулами воды и ионами OH? непрерывно происходит обмен протонами по уравнениям:
Н 3 O + + Н 2 O > Н 2 O + Н 3 O +
Н 2 O + OH? > OH? + Н 2 O
5. Электрохимические процессы
5.1. Электродные потенциалы. Гальванические элементы. ЭДС
При соприкосновении двух химически или физически разнородных материалов (металл 1 (проводник I рода) – металл 2 (проводник I рода), металл (проводник I рода) – раствор соли металла (проводник II рода), раствор электролита 1 (проводник II рода) – раствор электролита 2 (проводник II рода) и т. д.) между ними возникает двойной электрический слой (ДЭС). ДЭС является результатом упорядоченного распределения противоположно заряженных частиц на границе раздела фаз.
Образование ДЭС приводит к скачку потенциала?, который в условиях равновесия металл (проводник I рода) – раствор соли металла (проводник II рода) называется галъвани-потенциалом.
Система: металл (Me) – водный раствор соли данного Me – называется электродом или полуэлементом и схематически изображается следующим образом:
Электрод (п/э) записывается так, чтобы все вещества, находящиеся в растворе, были помещены слева, а электродный материал – справа от вертикальной черты.
? > 0, если на электроде протекает реакция восстановления Ме n+ + nе? - Ме 0 ,
? < 0, если на электроде протекает реакция окисления Ме 0 - Ме n+ + nе?.
Электродным потенциалом Е Ме n+ /Ме называется равновесная разность потенциалов, возникающая на границе фаз проводник I рода/проводник II рода и измеренная относительно стандартного водородного электрода.
уравнение Нернста, где n – число электронов, участвующих в электродной реакции; С Ме n+ – концентрация катионов; Е Ме n+ /Ме – стандартный электродный потенциал.
Контактный потенциал? ? – равновесный скачек потенциалов, возникающий на границе раздела двух проводников I рода.
Диффузионный потенциал? диф – равновесная разность потенциалов, возникающая на границе фаз проводник II рода/проводник II рода.
Гальванический элемент (г. э.) – электрическая цепь, состоящая из двух или нескольких п.э. и производящая электрическую энергию за счет протекающей в ней химической реакции, причем стадии окисления и восстановления химической реакции пространственно разделены.
Электрод, на котором при работе гальванического элемента протекает процесс окисления, называется анодом, электрод, на котором идет процесс восстановления, – катодом.
Правила ИЮПАК для записи гальванических элементов и реакций, протекающих в них1. В г. э. работа производится, поэтому ЭДС элемента считается величиной положительной.
2. Величина ЭДС гальванической цепи Е определяется алгебраической суммой скачков потенциала на границах раздела всех фаз, но так как на аноде протекает окисление, то ЭДС рассчитывают, вычитая из числового значения потенциала катода (правого электрода) значение потенциала анода (левого электрода) – правило правого полюса. Поэтому схему элемента записывают так, чтобы левый электрод был отрицательным (протекает окисление), а правый – положительным (протекает процесс восстановления).
3. Границу раздела между проводником I рода и проводником II рода обозначают одной чертой.
4. Границу между двумя проводниками II рода изображают пунктирной чертой.
5. Электролитный мостик на границе двух проводников II рода обозначают двумя пунктирными чертами.
6. Компоненты одной фазы записывают через запятую.
7. Уравнение электродной реакции записывают так, чтобы слева располагались вещества в окисленной форме (Ох), а справа – в восстановленной (Red).
Гальванический элемент Даниэля-Якоби состоит из цинковой и медной пластин, погруженных в соответствующие растворы ZnSO 4 и CuSO 4 , которые разделены солевым мостиком с раствором KCl: электролитический мостик обеспечивает электрическую проводимость между растворами, но препятствует их взаимной диффузии.
(-) Zn | Zn 2+ :: Cu 2+ | Cu (+)Реакции на электродах:
Zn 0 > Zn 2+ + 2e? Cu 2+ + 2е? > Cu 0Суммарный окислительно-восстановительный процесс:
Cu 2+ + Zn 0 > Cu 0 + Zn 2+Работа тока гальванического элемента (и, следовательно, разность потенциалов), будет максимальна при его обратимой работе, когда процессы на электродах протекают бесконечно медленно и сила тока в цепи бесконечно мала.
Максимальная разность потенциалов, возникающая при обратимой работе гальванического элемента, есть электродвижущая сила (ЭДС) гальванического элемента Е.
ЭДС элемента E Zn/ Cu = ? Cu 2+ /Cu + ? Zn 2+ /Zn + ? к + ? диф.
Без учета? диф и? к: E Zn / Cu = ? Cu 2+ /Cu + ? Zn 2+ /Zn = Е Cu 2+ /Cu + Е Zn 2+ /Zn – гальванические элементы, состоящие из двух одинаковых металлических электродов, опущенных в растворы соли этого металла с различными концентрациями С 1 > С 2 . Катодом в этом случае будет являться электрод с большей концентрацией, т. к. стандартные электродные потенциалы обоих электродов равны.

Концентрационные цепи

Единственным результатом работы концентрационного элемента является перенос ионов металла из более концентрированного раствора в менее концентрированный.
Работа электрического тока в концентрационном гальваническом элементе – это работа диффузионного процесса, который проводится обратимо в результате пространственного разделения его на два противоположных по направлению обратимых электродных процесса.
5.2. Классификация электродов
Электроды первого рода. Металлическая пластинка, погруженная в раствор соли того же металла. При обратимой работе элемента, в который включен электрод, на металлической пластинке идет процесс перехода катионов из металла в раствор либо из раствора в металл.
Электроды второго рода. Металл покрыт малорастворимой солью этого металла и находится в растворе, содержащем другую растворимую соль с тем же анионом. Электроды этого типа обратимы относительно аниона.
Электроды сравнения – электроды с точно известными и воспроизводимыми значениями потенциалов.
Водородный электрод представляет собой платиновую пластинку, омываемую газообразным водородом, погруженную в раствор, содержащий ионы водорода. Адсорбируемый платиной водород находится в равновесии с газообразным водородом.
Pt, Н 2 / Н +Электрохимическое равновесие на электроде:
2Н + + 2е? - Н 2 .Потенциал стандартного водородного электрода (с активностью ионов Н + 1 моль/л и давлением водорода 101,3 кПа) принят равным нулю.
Электродный потенциал нестандартного водородного электрода:

Каломельный электрод состоит из ртутного электрода, помещенного в раствор KCl, определенной концентрации и насыщенный каломелью Hg 2 Cl 2:
Hg / Hg 2 Cl 2 , KClКаломельный электрод обратим относительно анионов хлора
Хлорсеребряный электрод – обратим относительно анионов хлора:
Ag / AgCl, KClЕсли раствор KCl – насыщенный, то E AgC l = 0,2224 – 0,00065(t – 25), В.
Индикаторные электроды. Электроды, обратимые относительно иона водорода, используются на практике для определения активности этих ионов в растворе.
Хингидронный электрод представляет собой платиновую проволоку, опущенную в сосуд с исследуемым раствором, в который предварительно помещают избыточное количество хингидрона С 6 Н 4 O 2 С 6 Н 4 (OH) 2 – соединения хинона С 6 Н 4 O 2 и гидрохинона С 6 Н 4 (OH) 2 , способных к взаимопревращению в равновесном окислительно-восстановительном процессе, в котором участвуют ионы водорода:
С 6 Н 4 O 2 + 2H + + 2е? > С 6 Н 4 (OH) 2Наиболее часто употребляется стеклянный электрод в виде трубки, оканчивающейся тонкостенным стеклянным шариком. Шарик заполняется буферным раствором с определенным значением рН, в который погружен вспомогательный электрод (обычно хлорсеребряный). Для измерения рН стеклянный электрод погружают в исследуемый раствор в паре с электродом сравнения. Шарик стеклянного электрода предварительно обрабатывают в течение длительного времени раствором кислоты. При этом ионы водорода внедряются в стенки шарика, замещая катионы щелочного металла. Электродный процесс сводится к обмену ионами водорода между двумя фазами – исследуемым раствором и стеклом: Н р-р - Н ст + .
Стандартный потенциал Е ст 0 для каждого электрода имеет свою величину, которая со временем изменяется; поэтому стеклянный электрод перед каждым измерением рН калибруется по стандартным буферным растворам с точно известным рН.
Окислительно-восстановите льные электродыЭлектрод, состоящий из инертного проводника 1-го рода, помещенного в раствор электролита, содержащего один элемент в различных степенях окисления, называется окислительно-восстановительным или редокс-электродом.
Электродная реакция: Ох n+ + nе? - Red.
В данном случае инертный Me принимает косвенное участие в электродной реакции, являясь посредником передачи электронов от восстановленной формы Me (Red) к окисленной (Ох) или наоборот.

6. Поверхностные явления и адсорбция
6.1. Поверхностное натяжение и адсорбция по Гиббсу
Поверхностными явлениями называют процессы, происходящие на границе раздела фаз и обусловленные особенностями состава и строения поверхностного (пограничного) слоя.
G s = ?s,где G s – поверхностная энергия Гиббса системы, Дж; ? – коэффициент пропорциональности, называемый поверхностным натяжением, Дж/м 2 ; s – межфазная поверхность, м 2 .

Поверхностное натяжение о есть величина, измеряемая энергией Гиббса, приходящейся на единицу площади поверхностного слоя. Оно численно равно работе, которую необходимо совершить против сил межмолекулярного взаимодействия для образования единицы поверхности раздела фаз при постоянной температуре.

Из модели Дюпре, поверхностное натяжение равно силе, стремящейся уменьшить поверхность раздела и отнесенной к единице длины контура, ограничивающего поверхность

Способность растворенных веществ изменять поверхностное натяжение растворителя называется поверхностной активностью g:
Классификация веществ по влиянию на поверхностное натяжение растворителя1. Поверхностно-активные вещества (ПАВ) – понижают поверхностное натяжение растворителя (? р-р < ? 0) g > 0 (по отношению к воде – органические соединения дифильного строения).
2. Поверхностно-инактивные вещества (ПИВ) – незначительно повышают поверхностное натяжение растворителя (? р-р > ? 0) g < 0 (неорганические кислоты, основания, соли, глицерин, ?-аминокислоты и др).
3. Поверхностно-неактивные вещества (ПНВ) – практически не изменяют поверхностного натяжения растворителя (? р-р = ? 0) g = 0 (по отношению к воде веществами являются сахароза и ряд других).
Правило Дюкло-Траубе: в любом гомологическом ряду при малых концентрациях удлинение углеродной цепи на одну группу CH 2 увеличивает поверхностную активность в 3–3,5 раза:

Для водных растворов жирных кислот (уравнение Шишковского):
где b и К – эмпирические постоянные, b одинаково для всего гомологического ряда, К увеличивается для каждого последующего члена ряда в 3–3,5 раза.
Процесс самопроизвольного изменения концентрации какого-либо вещества у поверхности раздела двух фаз называется адсорбцией. Адсорбентом называется вещество, на поверхности которого происходит изменение концентрации другого вещества – адсорбата.
Изотерма адсорбции Гиббса:

Избыток адсорбата в поверхностном слое по сравнению с его первоначальным количествам в этом слое характеризует избыточную, или так называемую гиббсов-скую, адсорбцию (Г).
6.2. Адсорбция на границе твердое тело – газ
Физическая адсорбция возникает за счет ван-дер-ваальсовых взаимодействий адсорбированной молекулы с поверхностью, характеризуется обратимостью и уменьшением адсорбции при повышении температуры, т. е. экзотермичностью (тепловой эффект физической адсорбции обычно близок к теплоте сжижения адсорбата 10–80 кДж/моль).
Химическая адсорбция (хемосорбция) осуществляется путем химического взаимодействия молекул адсорбента и адсорбата, обычно необратима; является локализованной, т. е. молекулы адсорбата не могут перемещаться по поверхности адсорбента. Так как хемосорбция является химическим процессом, требующим энергии активации порядка 40-120 кДж/моль, повышение температуры способствует ее протеканию.
Уравнение Генри (мономолекулярная адсорбция на однородной поверхности при низких давлениях или малых концентрациях):
Г = Кс или Г = Кр,К – константа адсорбционного равновесия, зависящая от природы адсорбента и адсорбата; С, р – концентрация растворенного вещества или давление газа.
Теория мономолекулярной адсорбции Лэнгмюра
1. Адсорбция является локализованной и вызывается силами, близкими к химическим.
2. Адсорбция происходит на однородной поверхности адсорбента.
3. На поверхности может образоваться только один слой адсорбированных молекул.
4. Процесс адсорбции является обратимым и равновесным.
Изотерма адсорбции Лэнгмюра:

где Г 0 – емкость монослоя – константа, равная предельной адсорбции, наблюдаемой при относительно больших равновесных концентрациях, моль/м 2 ; b – константа, равная отношению константы скорости адсорбции и константе скорости десорбции.
Уравнение Фрейндлиха (адсорбция на неоднородной поверхности): Г = К Ф с n , где. К Ф – константа, численно равная адсорбции при равновесной концентрации, равной единице; n – константа, определяющая кривизну изотермы адсорбции (n = 0,1–0,6).
Молекулярная адсорбция из растворов:

где С 0 – исходная концентрация адсорба-та; С – равновесная концентрация адсорбата; V – объем раствора адсорбата; m – масса адсорбента.
Площадь S 0 , приходящаяся на одну молекулу в насыщенном адсорбционном слое, – посадочная площадка:

м 2 /молекула.
Толщина адсорбционного слоя:

где М – молекулярная масса ПАВ; ? – плотность ПАВ.
Правило Ребиндера: на полярных адсорбентах лучше адсорбируются полярные ад-сорбаты из малополярных растворителей; на полярных адсорбентах – неполярные адсорбаты из полярных растворителей.
Ориентация молекул ПАВ на поверхности адсорбента схематически изображена на рисунке:

6.3. Адсорбция из растворов электролитов
Обменная адсорбция – процесс обмена ионов между раствором и твердой фазой, при котором твердая фаза поглощает из раствора ионы какого-либо знака (катионы либо анионы) и вместо них может выделять в раствор эквивалентное число других ионов того же знака. Ввсегда специфична, т. е. для данного адсорбента к обмену способны только определенные ионы; обменная адсорбция обычно необратима.
Правило Пакета-Пескова-Фаянса: на поверхности кристаллического твердого тела из раствора электролита специфически адсорбируется ион, который способен достраивать его кристаллическую решетку или может образовывать с одним из ионов, входящих в состав кристалла, малорастворимое соединение.
7. Коллоидные (дисперсные) системы
Коллоидной (дисперсной) системой называется гетерогенная система, в которой одна из фаз представлена мелкими частицами, равномерно распределенными в объеме другой однородной фазы. Это ультрамикрогетерогенные системы, состоящие из частиц дисперсной фазы – совокупности раздробленных частиц, размер которых лежит в пределах 10 -9 -10 -5 м, и непрерывной дисперсионной среды, в которой распределены эти частицы.
Признаки коллоидного состояния вещества – дисперсность и гетерогенность.
Степень дисперсности? – величина, обратная среднему диаметру или, для несферических частиц, обратная среднему эквивалентному диаметру d (м -1):
Удельная поверхность – отношение общей площади поверхности дисперсной фазы S ДФ к ее общему объему или к ее массе:

7.1. Классификация и способы получения дисперсных систем
Классификация по агрегатному состоянию фаз


Дисперсной системы, у которой и дисперсная фаза, и дисперсионная среда являются газами, не существует, так как газы неограниченно растворимы друг в друге.
Классификация систем по размеру частиц дисперсной фазы:
1) высокодисперсные, 10 -9_ 10 -7 м (рубиновое стекло);
2) среднедисперсные, 10 -7_ 10 -5 м (растворимый кофе);
3) грубодисперсные, > 10 -5 м (капли дождя).
Способы получения коллоидных систем ДиспергированиеФизическое диспергирование: механическое измельчение с использованием коллоидных мельниц; электрическое распыление веществ; диспергирование ультразвуком и другие методы. Чтобы не дать образовавшимся частицам слипаться, диспергирование производят в присутствии стабилизатора – электролита или вещества, адсорбирующегося на границе раздела фаз (поверхностно-активные вещества).
Химическое диспергирование (пептизация): перевод в коллоидное состояние свежеприготовленного осадка с помощью пептизатора.
КонденсацияФизическая конденсация: 1) метод замены растворителя, который заключается в том, что в истинный раствор вещества добавляется смешивающаяся с растворителем жидкость, в которой само вещество малорастворимо; вследствие понижения растворимости вещества в новом растворителе раствор становится пересыщенным, и часть вещества конденсируется, образуя частицы дисперсной фазы; 2) метод конденсации из паров; исходное вещество находится в паре; при понижении температуры пар становится пересыщенным и частично конденсируется, образуя дисперсную фазу.
Химическая конденсация: любая химическая реакция, в результате которой образуется плохо растворимое соединение; чтобы при этом получить коллоидный раствор, реакцию необходимо вести в разбавленном растворе при небольшой скорости роста частиц, одно из исходных веществ берется в избытке и является стабилизатором.
7.2. Оптические свойства дисперсных систем
При падении света на дисперсную систему могут наблюдаться следующие явления:
прохождение света частицами дисперсной фазы (наблюдается для прозрачных систем, в которых частицы много меньше длины волны падающего света (r << ?);
преломление света частицами дисперсной фазы (если эти частицы прозрачны);
отражение света частицами дисперсной фазы (если частицы непрозрачны);
преломление и отражение света наблюдается для систем, в которых частицы много больше длины волны падающего света (r >> ?). Визуально это явление выражается в мутности этих систем;
рассеяние света наблюдается для систем, в которых частицы дисперсной фазы меньше, но соизмеримы с длиной волны падающего света (r ? 0,1 ?);
адсорбция (поглощение) света дисперсной фазой с превращением световой энергии в тепловую.
Уравнение Рэлея:

где I, I 0 – интенсивность рассеянного и падающего света; V – объем одной частицы; ? – частичная концентрация (число частиц в единице объема); ? – длина волны; n 1 , n 0 – показатели преломления частиц и среды соответственно.
Явление различной окраски коллоидного раствора в проходящем и рассеянном (отраженном) свете называется опалесценцией. В случае окрашенных растворов происходит наложение собственной окраски и окраски, вызванной опалесценцией (явление дихроизма света).
7.3. Молекулярно-кинетические свойства
Для коллоидных систем характерно броуновское движение – непрерывное беспорядочное движение частиц микроскопических и коллоидных размеров. Это движение тем интенсивнее, чем выше температура и чем меньше масса частицы и вязкость дисперсионной среды.
Диффузия – самопроизвольно протекающий процесс выравнивания концентрации частиц.
Закон Фика:

Вследствие большого размера коллоидных частиц диффузия в коллоидных системах замедленна по сравнению с истинными растворами.
Осмотическое давление:

где m общ – масса растворенного вещества; m – масса одной частицы; V – объем системы; N A – число Авогадро; Т – абсолютная температура; ? – частичная концентрация; k – постоянная Больцмана.
Для сферических частиц:

где? m – масса дисперсной фазы в единице объема раствора; ? – плотность дисперсионной среды; r – радиус частиц.
7.4. Строение мицеллы
Мицеллой лиофобной системы называется гетерогенная микросистема, которая состоит из микрокристалла дисперсной фазы, окруженного сольватированными ионами стабилизатора.
Потенциалопределяющими называются ионы, адсорбирующиеся на поверхности частички твердой фазы (агрегата) и придающие ей заряд. Агрегат вместе с потенциалопределяющими ионами составляет ядро мицеллы.
Противоионы – ионы, группирующиеся вблизи ядра мицеллы.
Расположение противоионов в дисперсионной среде определяется двумя противоположными факторами: тепловым движением (диффузией) и электростатическим притяжением.

Противоионы, входящие в состав плотного адсорбционного слоя, называются «связанными» и вместе с ядром составляют коллоидную частицу, или гранулу. Коллоидная частица (гранула) имеет заряд, знак которого обусловлен знаком заряда потенциалопределяющих ионов.
Противоионы, образущие диффузный слой, – «подвижные», или «свободные».
Коллоидная частица с окружающим ее диффузным слоем сольватированных про-тивоионов составляют мицеллу. В отличие от коллоидной частицы мицелла электронейтральна и не имеет строго определенных размеров.

В мицелле с ионным стабилизатором на границе раздела фаз имеется ДЭС, возникает разность потенциалов между дисперсной фазой и дисперсионной средой – термодинамический потенциал ф (межфазный), который определяется свойствами данной дисперсной системы, а также зарядом и концентрацией потенциалопределяющих ионов, адсорбированных на твердой фазе.

Перемещение заряженных коллоидных частиц в неподвижной жидкости к одному из электродов под действием внешнего электрического поля называется электрофорезом.
Поверхность, по которой происходит перемещение, называется поверхностью скольжения. Величина скачка потенциала на границе фаз, находящихся в движении относительно друг друга при электрофорезе и в броуновском движении, т. е. на поверхности скольжения, называется электрокинетическим или?-потенциалом (дзета-потенциал).
7.5. Устойчивость и коагуляция
Устойчивость дисперсных систем характеризует способность дисперсной фазы сохранять состояние равномерного распределения частиц во всем объеме дисперсионной среды.
Существует два вида относительной устойчивости дисперсных систем: седимента-ционная и агрегативная.
Седиментационная устойчивость – способность системы противостоять действию силы тяжести. Седиментация – это оседание частиц в растворе под действием силы тяжести.
Условие седиментационного равновесия: частица движется с постоянной скорость, т. е. равномерно, сила трения уравновешивает силу тяжести:
6??rU = 4/3?r 3 (? – ? 0)g,где? – плотность дисперсной фазы, ? 0 – плотность дисперсионной среды, g – ускорение силы тяжести, ? – вязкость среды.
Агрегативная устойчивость характеризует способность частиц дисперсной фазы противодействовать их слипанию между собой и тем самым сохранять свои размеры.
При нарушении агрегативной устойчивости происходит коагуляция – процесс слипания частиц с образованием крупных агрегатов. В результате коагуляции система теряет свою седиментационную устойчивость, т. к. частицы становятся слишком крупными и не могут участвовать в броуновском движении.
Причины коагуляции:
> изменение температуры;
> действие электрического и электромагнитного полей;
> действие видимого света;
> облучение элементарными частицами;
> механическое воздействие;
> добавление электролита и др.
Наибольший практический интерес вызывает коагуляция электролитами.
Виды коагуляции электролитамиКонцентрационная коагуляция наступает под действием индифферентных электролитов. Индифферентным называется электролит, при введении которого межфазный потенциал <р не изменяется. Данный электролит не содержит таких ионов, которые были бы способны к специфической адсорбции на частицах по правилу Па-нета-Фаянса, т. е. не способны достраивать кристаллическую решетку агрегата:

Состояние, при котором диффузный слой исчезнет и коллоидная частица становится электронейтральной, называется изоэлектрическим – электрокинетический потенциал (?) равен нулю, наступает коагуляция. Формула мицеллы в таком состоянии приобретает вид: {mnAg + nNO 3 ?} 0 .
Нейтрализационная коагуляция происходит при добавлению к золю неиндифферентного электролита. Неиндифферентным называется электролит, способный изменить межфазный (?) и линейно с ним связанный электрокинетический (?) потенциалы, т. е. данный электролит содержит ионы, способные специфически адсорбироваться на поверхности агрегата, достраивать его кристаллическую решетку или химически взаимодействовать с потенциалоп-ределяющими ионами.

Обратимый процесс, при котором коагулят вновь переходит в коллоидное состояние, называется пептизацией или дезагрегацией.
Правила коагуляции1. Все сильные электролиты, добавленные к золю в достаточном количестве, вызывают его коагуляцию. Минимальная концентрация электролита, вызывающая коагуляцию золя за определенный короткий промежуток времени, называется порогом коагуляции:

где С эл – концентрация электролита-коагулятора; V эл – объем добавленного электролита; V золя (обычно 10 мл) – объем золя.
2. Коагулирующим действием обладает тот ион, заряд которого совпадает по знаку с зарядом противоионов мицеллы лиофобного золя (заряд коагулирующего иона противоположен заряду коллоидной частицы). Этот ион называют ионом-коагулянтом.
3. Коагулирующая способность иона – коагулянта тем больше, чем больше заряд иона:
Правило значности:
? 1: ? 2: ? 3 = 1/1 6: 1/2 6: 1/3 6 = 729: 11: 1Коагулирующая способность иона при одинаковом заряде тем больше, чем больше его кристаллический радиус. Ag + > Cs + > Rb + > NH 4 + > K + > Na + > Li+ – лиотропный ряд.
Коллоидной защитой называется повышение агрегативной устойчивости золя путем введения в него ВМС (высокомолекулярное соединение) или ПАВ (поверхностно-активного вещества).
Защитным числом называется минимальное количество миллиграммов сухого вещества, которое необходимо для защиты 10 мл золя при добавлении к нему электролита в количестве, равном порогу коагуляции.
Начало физической химии было положено в середине XVIII века . Термин «Физическая химия», в современном понимании методологии науки и вопросов теории познания , принадлежит М. В. Ломоносову , который в впервые читал студентам Петербургского университета «Курс истинной физической химии». В преамбуле к этим лекциям он даёт такое определение: «Физическая химия - наука, которая должна на основании положений и опытов физических объяснить причину того, что происходит через химические операции в сложных телах». Учёный в трудах своей корпускулярно-кинетической теории тепла касается вопросов, в полной мере отвечающих вышеизложенным задачам и методам. Именно такой характер носят и экспериментальные действия, служащие подтверждению отдельных гипотез и положений настоящей концепции. М. В. Ломоносов следовал таким принципам во многих направлениях своих исследований: в разработке и практической реализации основанной им же «науки о стекле», в различных опытах, посвящённых подтверждению закона сохранения вещества и силы (движения); - в работах и экспериментах, имеющих отношение к учению о растворах - им была разработана обширная программа исследований настоящего физико-химического феномена, находящаяся в процессе развития до настоящего времени.
Затем последовал более чем столетний перерыв и одним из первых в России физикохимические исследования в конце 1850-х годов начал Д. И. Менделеев .
Следующий курс физической химии читал уже Н. Н. Бекетов в Харьковском университете в 1865 году .
Первая в России кафедра физической химии была открыта в 1914 году на физико-математическом факультете Санкт−Петербургского университета, осенью приступил к чтению обязательного курса и практическим занятиям по физической химии ученик Д. П. Коновалова М. С. Вревский .
Первый научный журнал, предназначенный для публикации статей по физической химии, был основан в 1887 году В. Оствальдом и Я. Вант-Гоффом .
Предмет изучения физической химии
Физическая химия является основным теоретическим фундаментом современной химии, использующим теоретические методы таких важнейших разделов физики, как квантовая механика , статистическая физика и термодинамика , нелинейная динамика , теория поля и др. Она включает учение о строении вещества, в том числе: о строении молекул, химическую термодинамику , химическую кинетику и катализ . В качестве отдельных разделов в физической химии выделяют также электрохимию , фотохимию , физическую химию поверхностных явлений (в том числе адсорбцию), радиационную химию , учение о коррозии металлов , физико-химию высокомолекулярных соединений (см. физика полимеров) и др. Весьма близко примыкают к физической химии и подчас рассматриваются как её самостоятельные разделы коллоидная химия , физико-химический анализ и квантовая химия . Большинство разделов физической химии имеет достаточно чёткие границы по объектам и методам исследования, по методологическим особенностям и используемому аппарату.
Различие между физической химией и химической физикой
Молекулярность элементарной реакции - число частиц, которые, согласно экспериментально установленному механизму реакции, участвуют в элементарном акте химического взаимодействия. Мономолекулярные реакции - реакции, в которых происходит химическое превращение одной молекулы (изомеризация, диссоциация и т. д.): H 2 S → H 2 + S Бимолекулярные реакции - реакции, элементарный акт которых осуществляется при столкновении двух частиц (одинаковых или различных): СН 3 Вr + КОН → СН 3 ОН + КВr Тримолекулярные реакции - реакции, элементарный акт которых осуществляется при столкновении трёх частиц: О 2 + NО + NО → 2NО 2 Реакции с молекулярностью более трёх неизвестны. Для элементарных реакций, проводимых при близких концентрациях исходных веществ, величины молекулярности и порядка реакции совпадают. Чётко определённой взаимосвязи между понятиями молекулярности и порядка реакции нет, так как порядок реакции характеризует кинетическое уравнение реакции, а молекулярность - механизм реакции. Основными направлениями химической термодинамики являются:
Химическая термодинамика тесно соприкасается с такими разделами химии , как
Физико-химический анализВ основе теории физико-химического анализа лежат сформулированные Н. С. Курнаковым принципы соответствия и непрерывности. Принцип непрерывности утверждает, что если в системе не образуются новые фазы или не исчезают существующие, то при непрерывном изменении параметров системы свойства отдельных фаз и свойства системы в целом изменяются непрерывно. Принцип соответствия утверждает, что каждому комплексу фаз соответствует определённый геометрический образ на диаграмме состав-свойство . Теория реакционной способности химических соединенийХимия высоких энергий (ХВЭ) изучает химические реакции и превращения, происходящие в веществе под воздействием нетепловой энергии. Механизмы и кинетика таких реакций и превращений характеризуются существенно неравновесными концентрациями быстрых, возбуждённых или ионизированных частиц с энергией большей, чем энергия их теплового движения и в ряде случаев химической связи. Носители нетепловой энергии, воздействующей на вещество: ускоренные электроны и ионы, быстрые и медленные нейтроны, альфа- и бета-частицы, позитроны, мюоны, пионы, атомы и молекулы при сверхзвуковых скоростях, кванты электромагнитного излучения, а также импульсные электрические, магнитные и акустические поля. Процессы химии высоких энергий различают по временны́м стадиям на физическую, протекающую за время фемтосекунд и менее, в течение которого нетепловая энергия распределяется в среде неравномерно и образуется «горячее пятно», физико-химическую, в течение которой проявляется неравновесность и негомогенность в «горячем пятне» и, наконец, химическую, в которой превращения вещества подчиняются законам общей химии. В результате образуются такие ионы и возбуждённые состояния атомов и молекул при комнатных температурах, которые не могут возникнуть за счёт равновесных процессов. Внешним проявлением ХВЭ служит образование ионов и возбуждённых состояний атомов и молекул при комнатных температурах, при которых эти частицы не могут возникнуть за счёт равновесных процессов. Н. Е. Аблесимов сформулировал релаксационный принцип управления свойствами неравновесных физико-химических систем. В случае, когда времена релаксации много больше длительности физического воздействия, существует возможность управления выходом химических форм, фаз и, как следствие, свойствами веществ (материалов), используя сведения о механизмах релаксации в неравновесных конденсированных системах на физико-химической стадии релаксационных процессов (в том числе и в процессе эксплуатации). Основные разделы ХВЭ
и другие. Лазерная химияИонизирующей способностью обладают электромагнитные излучения (рентгеновское излучение , γ-излучение , синхротронное излучение) и потоки ускоренных частиц (электронов , протонов , нейтронов , гелионов , тяжёлых ионов; осколки деления тяжёлых ядер и др.), энергия которых превышает потенциал ионизации атомов или молекул (в большинстве случаев, лежащий в пределах 10-15 эВ). В рамках радиационной химии рассматриваются некоторые химические процессы, невозможные при использовании традиционных химических подходов. Ионизирующие излучения могут сильно снижать температуру протекания химических реакций без применения катализаторов и инициаторов. История радиационной химии Радиационная химия возникла после открытия x-лучей В. Рентгеном в 1895 году и радиоактивности А. Беккерелем в 1896 году, которые первыми наблюдали радиационные эффекты в фотопластинках. Первые работы по радиационной химии были выполнены в 1899-1903 годах супругами М.Кюри и П. Кюри . В последующие годы наибольшее число исследований было посвящено радиолизу воды и водных растворов . Ядерная химияОсновные направления ядерной химии:
ЭлектрохимияЭлектрохи́мия - раздел химической науки, в котором рассматриваются системы и межфазные границы при протекании через них электрического тока , исследуются процессы в проводниках , на электродах (из металлов или полупроводников , включая графит) и в ионных проводниках (электролитах). Электрохимия исследует процессы |

3-е изд., испр. - М.: Высшая школа, 2001 - 512 с., 319 с.
Учебник составлен в соответствии с программой по физической химии.
В первой книге подробно изложены следующие разделы курса: квантовомеханические основы теории химической связи, строение атомов и молекул, спектральные методы исследования молекулярной структуры, феноменологическая и статистическая термодинамика, термодинамика растворов и фазовых равновесий.
Во второй части раздела курса физической химии электрохимия, химическая кинетика и катализ излагаются на основе представлений, развитых в первой части книги, - строение вещества и статистическая термодинамика. В разделе `Катализ` отражены кинетика гетерогенных и диффузионных процессов, термодинамика адсорбции и вопросы реакционной способности.
Для студентов вузов, обучающихся по химико-технологическим специальностям.
Книга 1.
Формат: djvu
Размер: 11,2 Мб
Скачать: drive.google
Книга 2.
Формат: djvu
Размер: 7 Мб
Скачать: drive.google
ОГЛАВЛЕНИЕ Книга 1.
Предисловие. 3
Введение 6
Раздел первый. Квантовомеханическое обоснование теории строения молекул и
химической связи
Г л а в а 1. Строение атома 9
§ 1.1. Квантовомеханические особенности микрочастиц 9
§ 1.2. Водородоподобный атом 11
§ 1.3. Атомные орбитали водородоподобного атома 14
§ 1.4. Спин электрона 21
§ 1.5. Многоэлектронные атомы 23
§ 1.6. Принцип Паули 26
§ 1.7. Электронные конфигурации атомов 28
Г л а в а 2. Молекулы. Теоретические методы, применяемые при изучении
строения молекул и химической связи 34
§ 2.1. Молекула. Потенциальная поверхность. Равновесная конфигурация 34
§ 2.2. Теория химической связи и ее задачи. Уравнение Шредингера для молекул 39
§ 2.3. Вариационный метод решения уравнения Шредингера 42
§ 2.4. Два основных метода теории строения молекул. Метод валентных связей и
метод молекулярных орбиталей 44
§ 2.5. Основные идеи метода молекулярных орбиталей 49
§ 2.6. Приближенное описание молекулярной орбитали в методе МО ЛКАО 50
§ 2.7. Молекула Щ в методе МО ЛКАО. Расчет энергии и волновой функции по
вариационному методу 53
§ 2.8. Молекула Н в методе МО ЛКАО. Ковалентная связь 58
Г л а в а 3. Двухатомные молекулы в методе МО ЛКАО 62
§ 3.1. Молекулярные орбитали гомонуклеарных двухатомных молекул 62
§ 3.2. Электронные конфигурации и свойства гомонуклеарных молекул, образованных
атомами элементов первого и второго периодов 65
§ 3.3. Гетеронуклеарные двухатомные молекулы 73
§ 3.4. Полярная связь. Электрический дипольный момент молекулы 78
§ 3.5. Насыщаемость ковалентной связи 81
§ 3.6. Донорно-акцепторная связь 82
§ 3.7. Ионная связь. Степень полярности химической связи 84
Г л а в а 4. Многоатомные молекулы в методе МО 88
§ 4.1. Молекулярные орбитали в многоатомных молекулах. Симметрия орбиталей.
Делокализованные и локализованные орбитали. Молекула НгО 88
§ 4.2. Описание молекулы метана. Делокализованные и локализованные МО.
Гибридизация орбиталей 95
§ 4.3. О предсказании равновесных конфигураций молекул 99
§ 4.4. Нежесткие молекулы 101
§ 4.5. Молекулы с кратными связями в методе МО ЛКАО 104
§ 4.6. Метод Хюккеля 108
§ 4.7. Описание ароматических систем в методе МОХ 110
§ 4.8. Химическая связь в координационных соединениях. Теория поля лигандов 117
§ 4.9. Ионная связь в кристалле 126
Г л а в а 5. Межмолекулярное взаимодействие 129
§ 5.1. Силы Ван-дер-Ваальса. Другие виды неспецифического взаимодействия 129
§ 5.2. Водородная связь 136
Раздел второй. Спектральные методы исследования строения и энергетических
состояний молекул
Г л а в а 6. Общие сведения о молекулярных спектрах. Элементы теории
молекулярных спектров 141
§ 6.1. Внутримолекулярное движение и электромагнитный спектр. 141
§ 6.2. Молекулярные спектры испускания, поглощения и комбинационного рассеяния.
Спектры ЭПР и ЯМР 145
§ 6.3. Вращательный спектр двухатомной молекулы (приближение жесткого ротатора)
150
§ 6.4. Колебательно-вращательный спектр двухатомной молекулы. Приближение
гармонического осциллятора 156
§ 6.5. Молекула - ангармонический осциллятор. Структура колебательного спектра
162
§ 6.6. Электронные спектры. Определение энергии диссоциации двухатомных молекул
169
§ 6.7. Вращательные спектры и строгие многоатомных молекул.... 171
§ 6.8. Колебания, спектр и строение многоатомных молекул 175
§ 6.9. Использование колебательных спектров для определения строения молекул 180
§ 6.10. Влияние межмолекулярного взаимодействия среды и агрегатного состояния на
колебательный спектр 183
Раздел третий. Химическая термодинамика
Г л а в а 7. Общие понятия. Первый закон термодинамики и его приложение 186
§ 7.1. Предмет и задачи химической термодинамики 186
§ 7.2. Основные понятия и определения химической термодинамики 188
§ 7.3. Первый закон термодинамики. Некруговые процессы 199
§ 7.4. Теплоемкость 202
§ 7.5. Влияние температуры на теплоемкость. Температурные ряды.. 208
§ 7.6. Квантовая теория теплоемкости кристаллического вещества 211
§ 7.7. Квантовостатистическая теория теплоемкости газообразного вещества 215
§ 7.8. Тепловые эффекты. Закон Гесса 217
§ 7.9. Применение закона Гесса к расчету тепловых эффектов 220
§ 7.10. Зависимость теплового эффекта от температуры. Уравнение Кирхгофа 227
Г л а в а 8. Второй закон термодинамики и ею приложение 235
§ 8.1. Самопроизвольные и несамопроизвольные процессы. Второй закон
термодинамики 235
§ 8.2. Энтропия 236
§ 8.3. Изменение энтропии в нестатических процессах 239
§ 8.4. Изменение энтропии как критерий направленности и равновесия в
изолированной «истеме 240
§ 8.5. Характеристические функции. Термодинамические потенциалы 241
§ 8.6. Критерии возможности самопроизвольного процесса и равновесия в закрытых
системах 249
§ 8.7. Изменение энтропии в некоторых процессах 251
§ 8.8. Энергия Гиббса смеси идеальных газов. Химический потенциал 261
§ 8.9. Общие условия химического равновесия 265
§ 8.10. Закон действующих масс. Константа равновесия для газофазных реакций 266
§ 8.11. Уравнение изотермы реакции 271
§ 8.12. Использование закона действующих масс для расчета состава равновесной
смеси 273
§ 8.13. Влияние температуры на химическое равновесие. Уравнение изобары реакции
282
§ 8.14. Интегральная форма зависимости изменения энергии Гиббса и константы
равновесия от температуры 284
§ 8.15. Химическое равновесие в гетерогенных системах 286
Г л а в а 9. Третий закон термодинамики и расчет химического равновесия 289
§ 9.1. Тепловая теорема Нернста. Третий закон термодинамики 289
§ 9.2. Расчет изменения стандартной энергии Гиббса и константы равновесия по
методу Темкина - Шварцмана 294
§ 9.3. Расчет изменения стандартной энергии Гиббса и константы равновесия с
помощью функций приведенной энергии Гиббса 297
§ 9.4. Адиабатические реакции 299
Г л а в а 10. Химическое равновесие в реальных системах 303
§ 10.1. Фугитивность и коэффициент фугитивности газов 303
§ 10.2. Расчет химического равновесия в реальной газовой системе при высоких
давлениях 312
§ 10.3. Расчет химического равновесия в системах, в которых одновременно
протекает несколько реакций 314
Г л а в а 11. Введение в статистическую термодинамику 320
§ 11.1. Статистическая физика и статистическая термодинамика.
Макроскопическое и микроскопическое описание состояния системы 320
§ 11.2. Микроскопическое описание состояния методом классической механики 323
§ 11.3. Микроскопическое описание состояния методом квантовой механики.
Квантовые статистики 324
§ 11.4. Два вида средних величин (микрокано -нические и канонические средние)
325
§ 11.5. Связь энтропии и статистического веса. Статистический характер второго
закона термодинамики 326
§ 11.6. Система в термостате. Каноническое распределение Гиббса. 330
§ 11.7. Сумма по состояниям системы и ее связь с энергией. Гельмгольца 335
§ 11.8. Сумма по состояниям частицы 337
§ 11.9. Выражение термодинамических функций через сумму по состояниям системы
340
§ 11.10. Сумма по состояниям системы одномерных гармонических осцилляторов.
Термодинамические свойства одноатомного твердого тела по теории Эйнштейна 343
§ 11.11. Квантовая статистика Больцмана. Закон Максвелла распределения молекул
по скоростям 346
§ 11.12. Статистики Ферми - Дирака и Бозе - Эйнштейна 352
§ 11.13.Общие формулы для вычисления термодинамических функций по молекулярным
данным 353
§ 11.14.Вычисление термодинамических функций идеального газа в предположении
жесткого вращения и гармонических колебаний молекул 357
Раздел четвертый. Растворы
Г л а в а 12. Общая характеристика растворов 365
§ 12.1. Классификация растворов 365
§ 12.2. Концентрация растворов 367
5 12.3. Специфика растворов. Роль межмолекулярного и химического взаимодействий,
понятие о сольватации 368
§ 12.4. Основные направления в развитии теории растворов 372
§ 12.5. Термодинамические условия образования растворов 374
§ 12.6. Парциальные молярные величины 375
§ 12.7. Основные методы определения парциальных молярных величин 379
§ 12.8. Парциальные и относительные парциальные молярные энтальпии 381
§ 12.9. Теплоты растворения и разбавления 382
§ 12.10.Термодинамические свойства идеальных жидких растворов 386
§ 12.11.3акон Рауля 390
§ 12.12. Температура кипения идеального раствора 392
§ 12.13.Температура замерзания идеального раствора 395
§ 12.14.0смотическое давление идеального раствора 397
§ 12.15.Неидеальные растворы 400
§ 12.16. Предельно разбавленные, регулярные и атермальные растворы 402
§ 12.17. Активность. Коэффициент активности. Стандартное состояние 404
§ 12.18.0смотический коэффициент 407
§ 12.19.Методы определения активностей 409
§ 12.20.Связь коэффициента активности и активности с термодинамическими
свойствами раствора и избыточные термодинамические функции 412
Раздел пятый.Фазовые равновесия
Г л а в а 13. Термодинамическая теория фазовых равновесий 415
§ 13.1. Основные понятия 415
§ 13.2. Условия фазового равновесия 418
§ 13.3. Правило фаз Гиббса 419
Глава 14. Однокомпонентные системы 421
§ 14.1. Применение правила фаз Гиббса к однокомпонентным системам 421
§ 14.2. Фазовые переходы первого и второго рода 422
§ 14.3. Уравнение Клапейрона - Клаузиуса 425
§ 14.4. Давление насыщенного пара 423
§ 14.5. Диаграммы состояния однокомпонентных систем 429
§ 14.6. Диаграмма состояния диоксида углерода 431
§ 14.7. Диаграмма состояния воды 432
§ 14.8. Диаграмма состояния серы 433
§ 14.9. Энантиотропные и монотропные фазовые переходы 435
Г л а в а 15. Двухкомпонентные системы 436
§ 15.1. Метод физико-химического анализа 436
§ 15.2. Применение правила фаз Гиббса к двухкомпонентным системам 437
§ 15.3. Равновесие газ - жидкий раствор в двухкомпонентных системах 438
§ 15.4. Равновесие жидкость - жидкость в двухкомпонентных системах 442
§ 15.5. Равновесие пар - жидкий раствор в двухкомпокентьых системах 444
§ 15.6. Физико-химические основы перегонки растворов 453
§ 15.7. Равновесие кристаллы - жидкий раствор в двухкомпонентных системах 457
§ 15.8. Равновесие жидкость - газ и кристаллы - газ (пар) в двухкомпонентных
системах 476
§ 15-9. Расчеты по диаграммам состояния 476
Г л а в а 16. Трехкомпонентные системы 482
§ 16.1. Применение правила фаз Гиббса к трехкомпонентным системам 482
§ 16.2. Графическое изображение состава трехкомпонентной системы 482
§ 16.3. Равновесие кристаллы - жидкий раствор в трехкомпонентных системах 484
§ 16.4. Равновесие жидкость - жидкость в трехкомпонентных системах 489
§ 16.5. Распределение растворяемого вещества между двумя жидкими фазами.
Экстракция 491
Приложение 495
Предметный указатель 497
ОГЛАВЛЕНИЕ Книга 2.
Предисловие 3
Раздел шестой. Электрохимия
Г л а в а 17. Растворы, электролитов 4
§ 17.1. Предмет электрохимии 4
§ 17.2. Специфика растворов электролитов 5
§ 17.3. Электролитическая диссоциация в растворе 6
§ 17.4. Средняя ионная активность и коэффициент активности 10
§ 17.5. Основные понятия электростатической теории сильных электролитов Дебая и
Хюккеля 13
§ 17.6. Основные понятия теории ассоциации ионов 22
§ 17.7. Термодинамические свойства ионов 24
§ 17.8. Термодинамика ионной сольватации 28
Г л а в а 18. Неравновесные явления в электролитах. Электрическая
проводимость электролитов 30
§ 18.1. Основные понятия. Законы Фарадея 30
§ 18.2. Движение ионов в электрическом поле. Числа переноса ионов. 32
§ 18.3. Электрическая проводимость электролитов. Удельная электрическая
проводимость 37
§ 18.4. Электрическая проводимость электролитов. Молярная электрическая
проводимость 39
§ 18.5. Молярная электрическая проводимость ионов гидроксония и гидроксида 43
§ 18.6. Электрическая проводимость неводных растворов 44
§ 18.7. Электрическая проводимость твердых и расплавленных электролитов 46
§ 18.8. Кондуктометрия 47
Г л а в а 19. Равновесные электродные процессы 49
§ 19.1. Основные понятия 49
§ 19.2. ЭДС электрохимической системы. Электродный потенциал 51
§ 19.3. Возникновение скачка потенциала на границе раствор-металл 53
§ 19.4. Диффузионный потенциал 55
§ 19.5. Строение двойного электрического слоя на границе раствор-металл 56
§ 19.6. Термодинамика обратимых электрохимических систем 60
§ 19.7. Классификация обратимых электродов 64
§ 19.8. Электродные потенциалы в неводных растворах 74
§ 19.9. Электрохимические цепи 75
§ 19.10. Применение теории электрохимических систем к изучению равновесия в
растворах 82
§ 19.11. Потенциометрия 85
Раздел седьмой. Кинетика химических реакций
Г л а в а 20. Законы химической кинетики 93
§ 20.1. Общие понятия и определения 93
§ 20.2. Скорость химической реакции 95
§ 20.3. Закон действующих масс и принцип независимости протекания реакций 101
Г л а в а 21. Кинетика химических реакций в закрытых системах. 105
§ 21.1. Односторонние реакции первого порядка 105
§ 21.2. Односторонние реакции второго порядка 109
§ 21.3. Односторонние реакции n-го порядка 111
§ 21.4. Методы определения порядка реакции 112
§ 21.5. Двусторонние реакции первого порядка 113
§ 21.6. Двусторонние реакции второго порядка 116
§ 21.Т. Параллельные односторонние реакции 117
§ 21.8. Односторонние последовательные реакции 119
§ 21.9. Метод квазистационарных концентраций 125
Г л а в а 22. Кинетика реакций в открытых системах 127
§ 22.1. Кинетика реакций в реакторе идеального смешения 127
§ 22.2. Кинетика реакций в реакторе идеального вытеснения 129
Г л а в а 23. Теория элементарного акта химического взаимодействия 133
§ 23.1. Элементарный химический акт 133
§ 23.2. Теория активных соударений 137
§ 23.3. Теория активированного комплекса 141
§ 23.4. Предэкспоненциальный множитель в уравнении Аррениуса по теории
переходного состояния 154
§ 23.5. Симметрия МО и энергия активации химических реакций 159
Г л а в а 24. Кинетика реакций в растворах, цепные и фотохимические реакции
166
§ 24.1. Особенности кинетики реакций в растворах 166
§ 24.2. Влияние среды на константу скорости реакции 170
§ 24.3. Кинетика ионных реакций в растворах 178
§ 24.4. Цепные реакции 181
§ 24.5. Фотохимические реакции 189
Г л а в а 25. Кинетика электродных процессов 196
§ 25.1. Скорость электрохимической реакции. Ток обмена 196
§ 25.2. Электродная поляризация 197
§ 25.3. Диффузионное перенапряжение 199
§ 25.4. Электрохимическое перенапряжение 205
§ 25.5. Другие виды перенапряжения 210
5 25.6. Температурно-кинетический метод определения природы поляризации при
электрохимических процессах 211
§ 25.7. Перенапряжение при электролитическом выделении водорода 213
§ 25.8. Электролиз. Напряжение разложения 217
§ 25.9. Поляризационные явления в химических источниках электрического тока 220
§ 25.10. Электрохимическая коррозия металлов. Пассивность металлов. Методы
защиты от коррозии 222
Раздел восьмой. Катализ
Г л а в а 26. Принципы каталитическою действия 228
§ 26.1. Основные понятия и определения 228
§ 26.2. Особенности кинетики каталитических реакций 232
§ 26.3. Энергия активации каталитических реакций 237
§ 26.4. Взаимодействие реагентов с катализатором и принципы каталитического
действия 241
Г л а в а 27. Гомогенный катализ 245
§ 27.1. Кислотно-основный катализ 246
§ 27.2. Окислительно-восстановительный катализ 255
§ 27.3. Ферментативный катализ 260
§ 27.4. Автокатализ, ингибирование и периодические каталитические реакции 266
§ 27.5. Применение в промышленности и перспективы развития гомогенного катализа
271
Г л а в а 28. Гетерогенный катализ. 273
§ 28.1. Структура поверхности гетерогенных катализаторов 273
§ 28.2. Адсорбция как стадия гетерогенно-каталитических реакций 277
§ 28.3. Механизм гетерогенно-каталитических реакций 282
§ 28.4. Кинетика гетерогенно-каталитических реакций на равнодоступной
поверхности 285
§ 28.5. Макрокинетика гетерогенно-каталитических процессов 292
§ 28.6. Применение гетерогенного катализа в промышленности 300
Литература 303
Приложение 305
Предметный указатель 312
Оглавление 316
1. Физическая химия: цель, задачи, методы исследования. Основные понятия физической химии.
Физ. химия - наука о закономерностях хим.процессов и хим. явлений.
Предмет физ.химии объяснение хим. явлений на основе более общих законов физики. Физ.химия рассматривает две основные группы вопросов:
1. Изучение строения и свойств вещества и составляющих его частиц;
2. Изучение процессов взаимодействия веществ.
Физ.химия ставит целью изучение связей м/у хим-ми и физ-ми явлениями. Знание таких связей необходимо для того, чтобы глубже изучить хим.реакции, протекающие в природе и используемые в технолог. процессах, управлять глубиной и направлением реакции. Основной целью дисциплины Физ.химия изучение общих связей и закономерностей хим. процессов, основанных на фундаментальных принципах физики. Физ.химия применяет физ. теории и методы к хим.явлениям.
Она объясняет ПОЧЕМУ и КАК происходят превращения веществ: хим. реакции и фазовые переходы. ПОЧЕМУ – хим.термодинамика. КАК- химическая кинетика.
Основные понятия физ.химии
Основной объект хим. термодинамики –это термодинамическая система. Термодинамич. система – любое тело или совокупность тел, способных обмениваться м/у собой и с др. телами энергией и в-вом. Системы подразделяют на открытые, закрытые и изолированные. Открыт ая - термодинамическая система обменивается с внешней средой и в-вом и энергией. Закрыт ая -система, в которой отсутствует обмен в-вом с окружающей средой, но она может обмениваться с ней энергией. Изолированн ая -система объем остается постоянным и лишена возможности обмениваться с окружающей средой и энергией и в-вом.
Система может быть гомогенной (однородной) или гетерогенной (неоднородной ). Фаза - это часть системы, которая в отсутствии внешнего поля сил обладает одинаковым составом во всех своих точках и одинаковыми термодинамич. св-вами и отделена от других частей системы поверхностью раздела. Фаза всегда однородна, т.е. гомогенна, поэтому однофазная система называется гомогенной. Система, состоящая из неск-ких фаз, называется гетерогенной.
Свойства системы подразделить на две группы: экстенсивные и интенсивные.
В термодинамике используются понятия равновесных и обратимых процессов. Равновесным –это процесс, проходящий через непрерывный ряд состояний равновесия. Обратимый термодинамический процесс – это процесс, который может быть проведен в обратном направлении без того, чтобы в системе и окружающей среде остались какие-либо изменения.
2. I-ый закон термодинамики. Внутренняя энергия, теплота, работа.
Первое начало термодинамики непосредственно связано с законом сохранения энергии. Исходя из этого закона, следует, что в любой изолированной системе запас энергии остается постоянным. Из законасохранения энергии вытекает еще одна формулировка первого начала термодинамики – невозможность создания вечного двигателя (perpetuum mobile) первого рода, который производил бы работу, не затрачивая на это энергии. Особенно важной для химической термодинамики формулировкой
первого начала является выражение его через понятие внутренней энергии: внутренняя энергия является функцией состояния, т.е. её изменение не зависит от пути процесса, а зависит только от начального и конечного состояния системы. Изменение внутренней энергии системы U может происходить за счет обмена теплотой Q и работой W с окружающей средой. Тогда из закона сохранения энергии следует, что полученная системой извне теплота Q расходуется на приращение внутренней энергии ΔU и работу W, совершенную системой, т.е. Q = ΔU +W . Данное у равнение является
математическим выражением первого начала термодинамики.
I начало термодинамики его формулировки:
в любой изолированной системе запас энергии остается постоянным;
разные формы энергии переходят друг в друга в строго эквивалентных количествах;
вечный двигатель (perpetuum mobile ) первого рода невозможен;
внутренняя энергия является функцией состояния, т.е. её изменение не зависит от пути процесса, а зависит только от начального и конечного состояния системы .
аналитическое выражение: Q = D U + W ; для бесконечно малого изменения величин d Q = dU + d W .
1-ое начало термодинамики устанавливает соотнош. м/у теплотой Q, работой А и изменением внутр. энергии системы ΔU. Изменение внутр. энергии системы равно кол-ву сообщенной системе теплоты минус кол-во работы, совершенной системой против внешних сил.
Уравнение (I.1)- математическая запись 1-го начала термодинамики, уравнение (I.2) – для бесконечно малого изменения сост. системы.
Внутр. энергия- функция сост.; это означает, что измен-е внутр. энергии ΔU не зависит от пути перехода системы из состояния 1 в состояние 2 и равно разности величин внутр. энергии U2 и U1 в этих состояниях: (I.3)
Внутр. энергия системы- это сумма потенциальной энергии взаимодейст. всех частиц тела м/у собой и кинетической энергии их движения (без учета кинетич. и потенциальн. энергий системы в целом). Внут. энергия системы зависит от природы в-ва, его массы и от параметров состоянии системы. Она возраст. с увеличением массы системы, так как является экстенсивным св-вом системы. Внутр. энергию обозначают литерой U и выражают в джоулях (Дж). В общем случае для системы с кол-вом в-ва 1 моль. Внутр. энергия, как и любое термодинамич. св-во системы, явл-ся функцией сост. Непосредственно в эксперименте проявляются только изменения внутр. энергии. Именно поэтому при расчетах всегда оперируют с её изменением U2 –U1 = U.
Все изменения внутр. энергии делятся на две группы. В 1-ую группу входит только 1-а форма перехода движения путем хаотических столкновений молекул двух соприкасающихся тел, т.е. путём теплопроводности (и одновременно путём излучения). Мерой передаваемого таким способом движения является теплота. Понятие теплоты связано с поведением огромного числа частиц – атомов, молекул, ионов. Они находятся в постоянном хаотическом (тепловом) движении. Теплота – форма передачи энергии. Второй способ обмена энергией – работа. Этот обмен энергии обусловлен действием, совершаемым системой, или действием, совершаемым над ней. Обычно работу обозначают символом W . Работа, также как и теплота, не является функцией состояния системы, поэтому величину, соответствующую бесконечно малой работе, обозначают символом частной производной - W .
Классификация наук основана на классификации форм движения материи и их взаимосвязи и различии. Поэтому для того, чтобы наметить границы физической химии с рядом разделов физики и химии, следует рассмотреть связь и различие между химической и физической формами движения.
Для химической формы движения, т. е. для химического процесса, характерно изменение числа и расположения атомов в молекуле реагирующих веществ. Среди многих физических форм движения (электромагнитное поле, движение и превращения элементарных частиц, физика атомных ядер и др.) особенно тесную связь с химическими процессами имеет внутримолекулярная форма движения (колебания в молекуле ее электронное возбуждение и ионизация). Простейший химический процесс - элементарный акт термической диссоциации молекулы имеет место при нарастании интенсивности (амплитуды и энергии) колебаний в молекуле, особенно колебаний ядер вдоль валентной связи между ними. Достижение известной критической величины энергии колебаний по направлению определенной связи в молекуле приводит к разрыву этой связи и диссоциации молекулы на две части.
Более сложные реакции с участием нескольких (обычно двух) молекул можно рассматривать как соединение двух молекул при их столкновении в непрочный и короткоживущий комплекс (так называемый активный комплекс) и быстро наступающее разрушение этого комплекса на новые молекулы, так как этот комплекс при внутренних колебаниях оказывается неустойчивым по определенным связям.
Таким образом, элементарный химический акт является особой, критической точкой колебательного движения молекул. Последнее само по себе не может считаться химическим движением, однако оно является основой для первичных химических процессов.
Для химического превращения значительных масс вещества, т. е. множества молекул, являются необходимыми столкновение молекул и обмен энергиями между ними (перенос энергии движения молекул продуктов реакции к молекулам исходных веществ путем столкновений). Таким образом, реальный химический процесс тесно связан и со второй физической формой движения - хаотическим движением молекул макроскопических тел, которое часто называют тепловым движением.
Выше намечены кратко и в самых общих чертах взаимные отношения химической формы движения с двумя физическими формами движения. Очевидно, имеются такие же связи химического процесса с излучением движения электромагнитного поля, с ионизацией атомов и молекул (электрохимия) и т.д.
Строение вещества . Этот раздел включает в себя строение атомов, строение молекул и учение об агрегатных состояниях.
Учение о строении атомов имеет большее отношение к физике, чем к физической химии. Это учение является основой для изучения строения молекул.
В учении о строении молекул исследуются геометрия молекул, внутримолекулярные движения и силы, связывающие атомы в молекуле. В экспериментальных исследованиях строения молекул наибольшее применение получил метод молекулярной спектроскопии (включая радиоспектроскопию), широко используются также электрические, рентгенографические, магнитные и другие методы.
В учении об агрегатных состояниях рассматриваются взаимодействия молекул в газах, жидкостях и кристаллах, а также свойства веществ в различных агрегатных состояниях. Этот очень важный для физической химии раздел науки может считаться частью физики (молекулярная физика).
Весь раздел о строении вещества может рассматриваться также как часть физики.
Химическая термодинамика . В этом разделе на основе законов общей термодинамики излагаются законы химического равновесия и учение о фазовых равновесиях, которое обычно называют правилом фаз. Частью химической термодинамики является термохимия, в которой рассматриваются тепловые эффекты химических реакций.
Учение о растворах ставит своей целью объяснение и предсказание свойств растворов (гомогенных смесей нескольких веществ) на основании свойств веществ, составляющих раствор.
Решение этой задачи требует построения общей теории взаимодействия разнородных молекул, т. е. решения основной задачи, молекулярной физики. Для развития общей теории и частных обобщений изучаются молекулярная структура растворов и различные их свойства в зависимости от состава.
Учение о поверхностных явлениях . Изучаются разнообразные свойства поверхностных слоев твердых тел и жидкостей (границы раздела между фазами); одно из основных изучаемых явлений в поверхностных слоях - это адсорбция (накопление веществ в поверхностном слое).
В системах, где поверхности раздела между жидкими, твердыми и газообразными фазами сильно развиты (коллоидные растворы, эмульсии, туманы, дымы), свойства поверхностных слоев приобретают основное значение и определяют многие своеобразные свойства всей системы в целом. Такие микрогетерогенные системы изучаются коллоидной химией, которая является крупным самостоятельным разделом физической химии и самостоятельной учебной дисциплиной в химических высших учебных заведениях.
Электрохимия. Изучается взаимодействие электрических явлений и химических реакций (электролиз, химические источники электрического тока, теория электросинтеза). В электрохимию включают обычно учение о свойствах растворов электролитов, которое с равным правом можно отнести и к учению о растворах.
Химическая кинетика и катализ . Изучается скорость химических реакций, зависимость скорости реакции от внешних условий (давление, температура, электрический разряд и др.), связь скорости реакции со строением и энергетическими состояниями молекул, влияние на скорость реакции веществ, не участвующих в стехиометрическом уравнении реакции (катализ).
Фотохимия. Исследуется взаимодействие излучения и веществ, участвующих в химических превращениях (реакции, протекающие под влиянием излучения, например фотографические процессы и фотосинтез, люминесценция). Фотохимия тесно связана с химической кинетикой и учением о строении молекул.
Приведенный перечень основных разделов физической химии не охватывает некоторых недавно возникших областей и более мелких разделов этой науки, которые можно рассматривать как части более крупных разделов или как самостоятельные разделы физической химии. Таковы, например, радиационная химия, физико-химия высокомолекулярных веществ, магнетохимия, газовая электрохимия и другие разделы физической химии. Значение некоторых из них в настоящее время быстро растет.
Методы физико-химического исследования
Основные методы физической химии, естественно, являются методами физики и химии. Это - прежде всего экспериментальный метод - исследование зависимости свойств веществ от внешних условий и экспериментальное изучение законов протекания химических реакций во времени и законов химического равновесия.
Теоретическое осмысливание экспериментального материала и создание стройной системы знаний свойств веществ и законов химических реакций основано на следующих методах теоретической физики.
Квантово-механический метод (в частности, метод волновой механики), лежащий в основе учения о строении и свойствах отдельных атомов и молекул и взаимодействии их между собой. Факты, относящиеся к свойствам отдельных молекул, получаются, главным образом, с помощью экспериментальных оптических методов.
Метод статистической физики , дающий возможность рассчитать свойства вещества; состоящего из множества молекул («макроскопические» свойства), на основании сведений о свойствах отдельных молекул.
Термодинамический метод , позволяющий количественно связывать различные свойства вещества («макроскопические» свойства) и рассчитывать одни из этих свойств на основании опытных величин других свойств.
Современные физико-химические исследования в любой конкретной области характеризуются применением разнообразных экспериментальных и теоретических методов для изучения различных свойств веществ и выяснения их связи со строением молекул. Вся совокупность данных и указанные выше теоретические методы используются для достижения основной цели - выяснения зависимости направления, скорости и пределов протекания химических превращений от внешних условий и от строения молекул - участников химических реакций.